
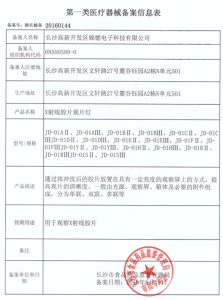
第一类医疗器械备案产品检验报告如何办理?厂家自检报告是否可以备案
解答:关于第一类医疗器械在备案的过程中,是否可以使用厂家自己的检测报告问题。根据
中华人民共和国国务院令 第680号
国务院关于修改《医疗器械监督管理条例》的决定
第十条 第一类医疗器械产品备案,由备案人向所在地设区的市级人民政府食品药品监督管理部门提交备案资料。其中,产品检验报告可以是备案人的自检报告;临床评价资料不包括临床试验报告,可以是通过文献、同类产品临床使用获得的数据证明该医疗器械安全、有效的资料。
根据法令,是可以提供自检报告的。
第六条 国家对医疗器械按照风险程度实行分类管理:
第一类是风险程度低,实行常规管理可以保证其安全、有效的医疗器械。
第二类是具有中度风险,需要严格控制管理以保证其安全、有效的医疗器械。
第三类是具有较高风险,需要采取特别措施严格控制管理以保证其安全、有效的医疗器械。
评价医疗器械风险程度,应当考虑医疗器械的预期目的、结构特征、使用方法等因素。
国务院药品监督管理部门负责制定医疗器械的分类规则和分类目录,并根据医疗器械生产、经营、使用情况,及时对医疗器械的风险变化进行分析、评价,对分类规则和分类目录进行调整。制定、调整分类规则和分类目录,应当充分听取医疗器械注册人、备案人、生产经营企业以及使用单位、行业组织的意见,并参考国际医疗器械分类实践。医疗器械分类规则和分类目录应当向社会公布。
第七条 医疗器械产品应当符合医疗器械强制性国家标准;尚无强制性国家标准的,应当符合医疗器械强制性行业标准。